Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2¶
作者 · Love, Huber & Anders, 期刊 · Genome Biology, 年份 · 2014, DOI · https://doi.org/10.1186/s13059-014-0550-8
一句话:DESeq2 把 RNA-seq 差异表达从“看 fold change 和 p 值”推进到负二项 GLM、dispersion shrinkage 和 LFC shrinkage 的稳定推断框架。
1. 背景与前问¶
RNA-seq 早期很快遇到一个问题:reads count 看似简单,但基因之间表达量差异巨大,样本之间 library size 不同,生物重复之间 variance 明显大于 Poisson。若直接对 normalized counts 做 t-test,会同时错过 count discreteness、mean-variance relationship 和 multiple testing。
DESeq、edgeR 已经建立了 negative binomial(负二项)框架,但真实项目里还有三个难点。第一,小样本下每个基因单独估 dispersion 极不稳定。第二,高噪音低 count 基因会产生夸大的 fold change。第三,复杂 design 需要 GLM,而不是简单两组比较。DESeq2 的目标就是把这些问题系统化。
2. 核心问题¶
核心问题一句话:如何在小样本 RNA-seq 中稳定估计差异表达,同时避免 dispersion 和 log2 fold change 被噪音主导?
这不是单纯“换一个工具”。DESeq2 的设计思想是经验贝叶斯 shrinkage:每个基因有自己的信息,但也要向全局 mean-dispersion trend 和 fold-change prior 借力。这样做牺牲一点自由度,换取更稳定的推断。
3. 实验设计的关键决策¶
DESeq2 面向的是最常见但最困难的生物学场景:每组 2-5 个生物重复、上万个基因、多因素 design、count depth 不一。作者没有假设大样本渐近条件完全可靠,而是围绕 small-n RNA-seq 设计 shrinkage。
它选择 gene-level raw counts 作为输入,而不是 TPM/FPKM。原因是 NB GLM 需要整数 count 来建模 sampling + biological variation;长度校正后的连续量不再服从同一 count likelihood。这个设计取舍是 DESeq2 的核心,不是软件偏好。
4. 数据生成与处理¶
DESeq2 的模型可以写成:
其中 \(K_{ij}\) 是基因 \(i\) 在样本 \(j\) 的 count,\(\mu_{ij}\) 是均值,\(\alpha_i\) 是基因特异 dispersion。均值分解为:
\(s_j\) 是 size factor,校正 library size 和 composition;\(x_j\) 是 design matrix;\(\beta_i\) 是基因 \(i\) 的回归系数。负二项方差为:
推导逻辑来自 Poisson-Gamma mixture:如果测序抽样近似 Poisson,而真实表达均值在生物重复间按 Gamma 波动,边缘分布就是 NB,方差自然多出 \(\alpha\mu^2\) 项。
DESeq2 的关键不是写出 NB,而是三层 moderation:
- 先估每个基因的 dispersion。
- 拟合 mean-dispersion trend。
- 将基因 dispersion 向 trend shrink,同时保留真正高变异基因。
LFC shrinkage 则针对低 count 基因的巨大 log2 fold change。低表达基因只要一个样本出现少数 reads,就可能产生很大 naive LFC;DESeq2 用 prior 把不稳定估计拉回更保守的位置。
5. 关键 Figure 拆解¶
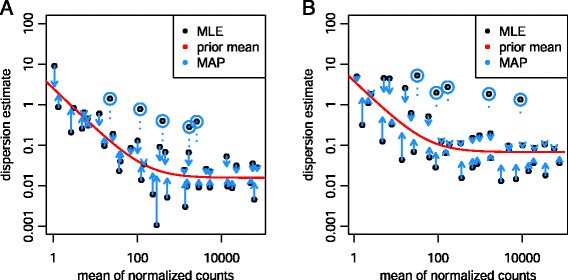
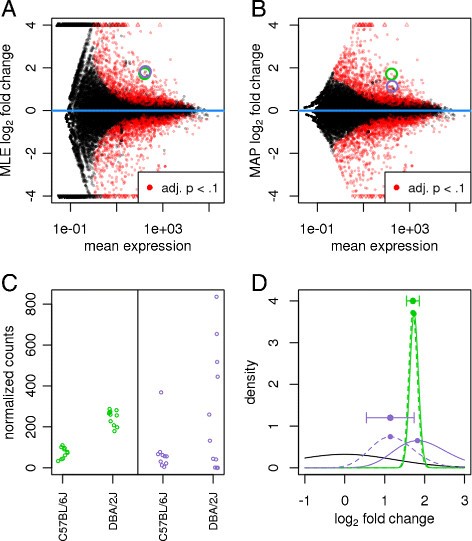
Figure 1:workflow 与模型结构¶
这张图不是装饰,而是 DESeq2 的统计路线图:raw counts -> size factors -> dispersion estimation -> GLM fitting -> Wald/LRT -> multiple testing。它的生物学声明是:差异表达不是“先归一化再比较均值”,而是在 count likelihood 中同时处理 normalization、variance 和 design。
实际科研读法:如果你的 design 有 batch、genotype、treatment、interaction,DESeq2 的价值在 design matrix。错误 design 比错误工具更危险。
Figure 2:dispersion shrinkage¶
这张图展示每基因 dispersion 估计、fitted trend 和 shrinked estimates。统计声明是:单个基因的 dispersion MLE 噪音很大,尤其小样本;把它向全局 trend 收缩可以降低方差。
这张图支撑的生物学意义很直接:如果 dispersion 估太小,p 值会过度乐观,假阳性上升;如果估太大,真差异被淹没。DESeq2 的 moderation 是在 false positive 和 power 之间做稳定折中。
Figure 3/4:LFC shrinkage 与方法比较¶
这些图展示 shrinkage 后的 fold change 更稳定,尤其低 count 基因不再满屏极端 LFC。生物学声明是:排名和解释 DEG 时,effect size 必须考虑估计不确定性。一个低 count 基因的 16-fold change 未必比高 count 基因的 1.5-fold change 更可信。
这对写论文尤其重要。DESeq2 的 adjusted p 值用于显著性控制;shrunken LFC 更适合排序、可视化和生物学解释。
6. 结论的强度边界¶
强支持的结论:NB GLM 是 bulk RNA-seq count 的合理工作模型;dispersion shrinkage 能改善小样本稳定性;LFC shrinkage 能减少低 count 基因的夸大效应;复杂 design 应该在模型中处理,而不是靠手工分组。
强烈暗示但不是绝对的结论:DESeq2 在所有 RNA-seq 场景都最优。实际不是。极端 composition bias、single-cell pseudo-bulk、transcript-level uncertainty、zero inflation 或强 outlier 都可能需要额外方法或诊断。
常见误读:把 DESeq2 normalized counts 导出后再做 t-test;把 shrunken LFC 当作原始 fold change;用 TPM 输入 DESeq2;忽视 batch 完全混杂。这些不是小错,而是改变统计问题。
7. 如果今天重做¶
2026 年看 DESeq2,我会保留它作为 gene-level bulk RNA-seq 的核心框架,但在项目中做这些升级:
- 上游用 Salmon/kallisto + tximport 时,明确 gene-level count construction 和 length scaling。
- 对复杂设计,预先写 design matrix,检查 rank 和 confounding。
- 对细胞组成混合组织,加入 deconvolution 或直接做 cell-type-specific/pseudobulk。
- 对 LFC shrinkage,用 apeglm/ashr 这类后续方法,而不是只停留在早期 normal prior。
- 对结果解释,报告 independent filtering、Cook's distance/outlier 处理、dispersion plot、MA plot 和 PCA。
植物项目还要额外注意:多倍体 homeolog mapping 会改变 count;强处理可能造成全局表达 shift,使 size factor 假设受压;组织样本常有细胞组成和发育阶段混合。DESeq2 能处理统计离散度,但不能修复设计混杂。
8. 我学到了什么¶
(Peter 填)
横向连接¶
- [[_concepts/poisson-vs-negative-binomial]]
- [[_concepts/glm-unified-view]]
- [[03-bulk-RNAseq/why-negative-binomial-not-poisson]]
- [[03-bulk-RNAseq/deseq2-dispersion-three-steps]]
- [[03-bulk-RNAseq/lfc-shrinkage-priors]]
- [[03-bulk-RNAseq/_papers/mortazavi-2008-nature-methods-rnaseq]]
参考¶
- Love et al. (2014), Genome Biology, DOI: https://doi.org/10.1186/s13059-014-0550-8
- Anders & Huber (2010), Genome Biology, DOI: https://doi.org/10.1186/gb-2010-11-10-r106
- Robinson et al. (2010), Bioinformatics, DOI: https://doi.org/10.1093/bioinformatics/btp616
- McCarthy et al. (2012), Nucleic Acids Research, DOI: https://doi.org/10.1093/nar/gks042
- Zhu et al. (2019), Bioinformatics — apeglm LFC shrinkage